
表观遗传学是一个迅速发展的癌症研究领域。本页讨论了表观遗传学的一些基础知识以及表观遗传变化与癌症之间的联系。
- 表观遗传学的介绍
- 表观遗传修饰
- 基因外的表观遗传变化
- 突变原因
- 癌症中DNA甲基化的变化
- 癌症中蛋白质的修饰的变化
- 表观遗传学与肿瘤转移
- 表观遗传学与癌症预防
- 表观遗传学与癌症检测
- 表观遗传蛋白作为癌症生物标志物
- 表观遗传学作为抗癌药物的靶点
表观遗传学概论
癌症的生物学基础在研究人员发现了疾病和缺陷基因之间的联系之前是未知的。他们发现在特定基因的DNA序列中的变化(突变)导致了癌症中不受控制的细胞繁殖(细胞分裂)。1 这导致了两大类与癌症有关的基因的发现。首先发现的是癌基因,当它们的活性增加时便会致癌,后来又发现了抑癌基因。肿瘤抑制物通常能阻断癌症,但当它们被改变或者消除时,却能帮助癌症的产生。 最近,人们认识到癌症也可能是表突变的结果,即是在不改变DNA序列的情况下微小的化学变化改变了基因的活性。
几乎我们身体中的每一个细胞都含有相同的DNA序列,但很快就可以清楚地看到,我们所有的细胞看起来和行为都不一样。心脏细胞的外观和行为都与肺细胞大有不同,尽管两种细胞都含有相同的DNA。表观遗传学解释了在不同细胞中观察到各种活动的原因。Epi-是“上面”的希腊语前缀。 表观遗传变化为细胞控制和调节基因活性提供了一种不必永久改变基因的途径。表观遗传反而控制依赖于组成染色体的DNA和蛋白质的微小,可逆的变化。要了解表观遗传学,就必须了解DNA的本质。DNA由四种化学单元组成(核苷酸)。 这些核苷酸彼此共享一些部分,但它们都有独特的部分,称为“碱基”--DNA中的碱基是鸟嘌呤(G),胞嘧啶(C)、腺嘌呤(A)和胸腺嘧啶(T)。 DNA是螺旋状的阶梯状结构,中间有一对对的碱基。在细胞中,DNA没有办法自由漂浮;它被紧紧地包裹在染色体中--DNA紧紧地包裹在称为组蛋白的蛋白质上组成的结构。2 DNA以这种方式包裹有几种原因。一是DNA分子非常非常长,如果都是未卷曲的,单个细胞中的DNA大约有6英尺(2米)长!3 如果没有良好的组织,几乎不可能容纳到一个细胞里。另外一个原因是严格控制基因活性,使基因只有在需要时才被激活。
那么,一个细胞如何启动或激活它所需要的基因呢?首先,含有基因的DNA部分需要解开,而细胞通过修改组蛋白来完成这一任务。酶通过添加或移除小的化学标记来让组蛋白放松对DNA的控制。当DNA变得可用时,不同的蛋白质就能够粘在目标基因上并使用编码的信息。这一过程受到严格控制,因为细胞中不受控制的基因活动能够引起多种问题,其中也包括癌症的发展。
表观遗传修饰
如上所诉,表观遗传控制依赖于DNA或者染色体中蛋白质的微小化学变化。表观遗传修饰有几种类型。主要种类如下所诉。
1.DNA修饰
最常见的DNA修饰是甲基化。甲基化是在特定的碱基上加上一个叫做甲基的小化学基因(-CH3),这些碱基被称为DNA甲基转移酶(DNMTs)的酶修饰。最常见的碱基是胞嘧啶。在一个基因中加入许多甲基通常会导致基因失活(沉默)。有几种方法可以使这些小群体的加入导致一个基因的关闭。第一种是DNA上的甲基会阻止读取DNA的酶去识别并与目标基因结合。第二种是DNA上的甲基会招募阻止基因激活的DNA结合蛋白。如果正确的蛋白质不能粘附在基因上,基因就无法被“读取”。DNA甲基化,就像组蛋白质修饰一样被严格控制着,当正常的DNA甲基化模式被改变时,就会出现问题。一些肿瘤细胞已经被发现在许多基因上大面积缺少DNA甲基化。因为甲基化与基因活性有关,所以单个基因甲基化的变化可以导致癌症。驱动细胞繁殖的基因可以变得过于活跃,或者通常阻止细胞生长失控的基因也可以被关闭。

DNA包裹在组蛋白的周围。每组8个组蛋白成为核小体。
2. 组蛋白修饰
DNA 是以一种叫做核小体的结构组织起来的,类似于一根绳子上的珠子。每个核小体由包裹在称为蛋白的蛋白质上的DNA组成,每个核小体中有8个组蛋白(每个组蛋白H2A,H2B,H3和H4都有两个拷贝)。2 组蛋白排列在核小体中,因此蛋白质的末端(它们的“尾巴”)从结构的中心突出。蛋白质尾部都是组蛋白修饰的主要部位。 L和所有蛋白质一样,组蛋白也是由氨基酸组成的。位于组蛋白尾部的氨基酸是附加或去除化学标记的酶的目标。赖氨酸和丝氨酸是常见的修饰目标。
与DNA甲基化相比,组蛋白修饰是一个相对复杂的过程。DNA甲基化只涉及到两种酶,一种是加甲基的胞嘧啶,另一种是去除甲基胞嘧啶。而组蛋白有三种不同的修饰类型。组蛋白修饰的三种类型分别为:
- 组蛋白磷酸化: 在组蛋白的氨基酸中加入磷酸基。
- 组蛋白甲基化:三种类型中最复杂的类型。甲基会被加到含有氨基酸的组蛋白里。一个或者更多的氨基酸可以被修饰,而个别氨基酸也可以被加入多个甲基。这些步骤的复杂性体现在与组蛋白甲基化相关的许多酶中。每次添加或者去除都需要一种特定的酶。
- 组蛋白乙酰化.4 在氨基酸乃氨酸上加上或者去掉一个乙酰基。常见于组蛋白的尾部。组蛋白乙酰化使组蛋白带较少的正电荷,组蛋白与其结合DNA之间的吸引力减弱。这导致了基因活性的增加,因为DNA更加松散,更有效。
上面描述的微小变化改变了核小体的形状,改变了DNA于特定“珠子”相互作用的方式。组蛋白修饰是一个非常复杂的过程,受到严格控制。当组蛋白修饰模式改变时,它可以导致不受调节的细胞活性或者使基因沉默。最终这会导致细胞死亡,或者更严重的可以导致癌症。
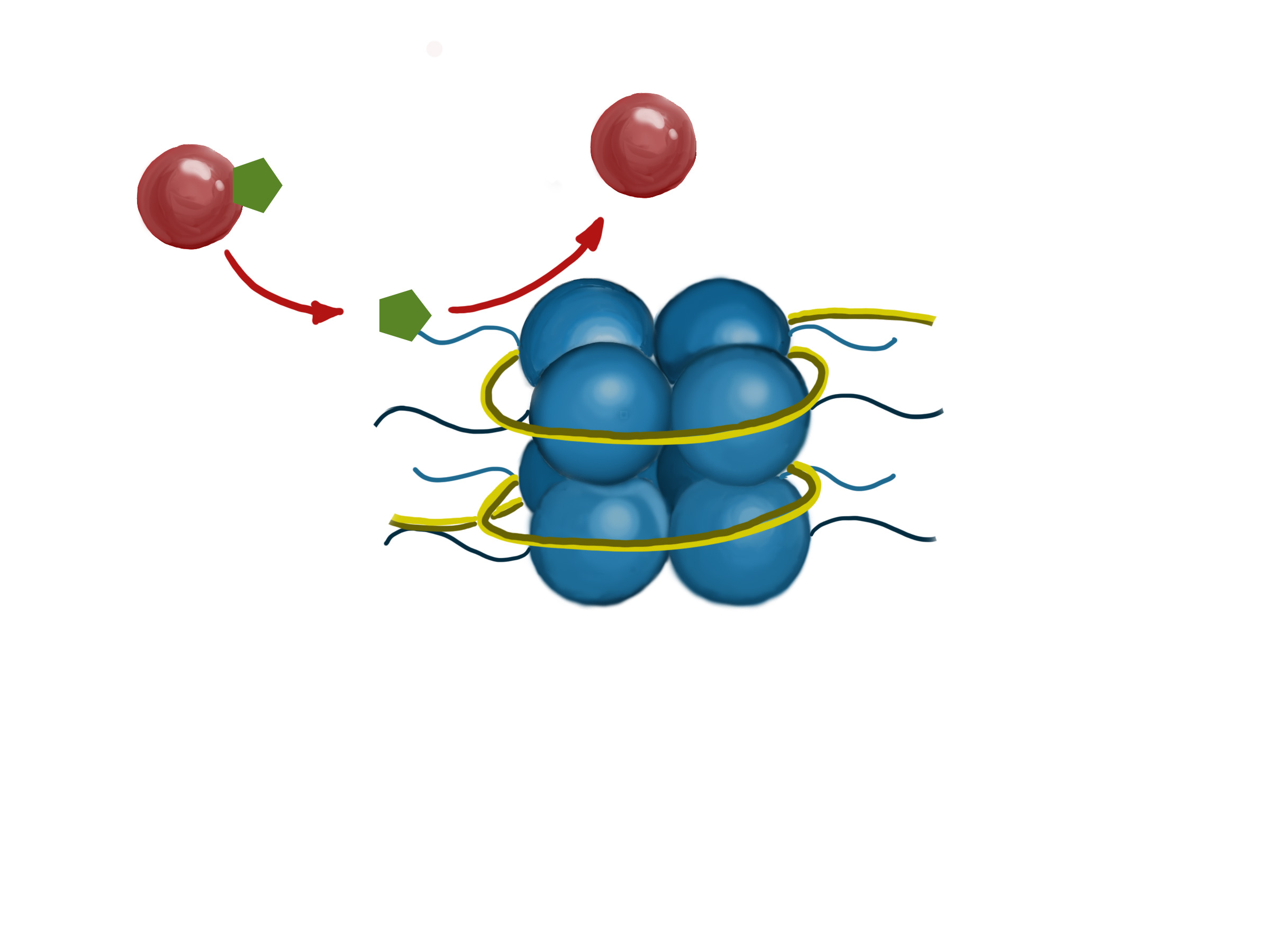
图中显示了一种酶(红球)在组蛋白的尾部添加了一个化学基因(绿色六边形)。
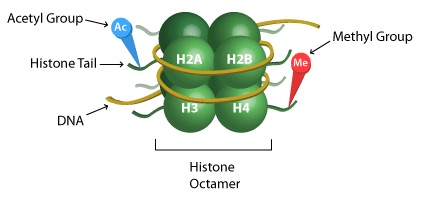
核小体结构更详细的图片,显示在组蛋白“尾部”上甲基化和乙酰化的目标。
基因外的表观遗传变化
现在人们已经了解了表观遗传学如何导致癌症(致癌)的发展。如上所诉,特定癌基因和抑癌基因的异常表观遗传修饰可导致细胞生长和分裂失控。然而,基因外DNA区域的异常表观遗传修饰也可能导致癌症。蛋白质编码基因构成了我们DNA的一小部分。人类有大约20000个基因,我们的基因所占的DNA不到我们DNA的10%。5 剩下的90%是由非编码序列组成的。非编码序列包括调节序列和负责管理染色体结构的DNA区域。 这些区域的变化与包括癌症在内的集中疾病相关。值得注意的是,发生在一个细胞中的表突变会传递给当该细胞通过丝分裂繁殖时形成的子细胞。
表突变的原因
什么会导致异常的表观遗传修饰? 环境和人类行为是主要原因。不良的饮食,缺乏锻炼,药物,暴露于环境化学物质或辐射中等,这些都有可能导致表观突变。例如,吸烟已经被证明会影响多个器官系统的DNA甲基化模式。这些受影响的基因与癌症,骨质疏松症,慢性阻塞性疾病(COPD),心血管疾病,和类风湿6除了行为和环境之外,一个人本身拥有的一套基因肯定也是一部分的原因。
关于表观遗传学一个非常重要的事实就是基因的变化可以从父母传递给子女。这个术语是“跨代遗传”。最早的研究表明这种跨代表观遗传是在小鼠身上进行的。在这项研究中,父母在特定气味中生活会影响后代的行为和感觉神经元。7 这项研究是开创性的,因为它表明了我们可以被父母和祖父母的经历所影响。这背后确切的方法还仍不清楚。
跨代遗传被认为是不可能是因为一个叫做生殖系重组的一个过程。8 在这个过程中,复制细胞(精子和卵子细胞)的DNA被“重置”,所有的表观遗传标记和修饰都被清除。理论上,这将消除表观遗传事件在世代之间转移的可能性。不过,这个过程并不像我们先前想象的那样完整。研究人员鉴定了生殖细胞中的DNA甲基化点,发现尽管大部分基因组确实去甲基化,但仍有相当大数量的基因保留了它们的表观遗传标记。8 发生这种情况的原因还有待讨论,但是一些研究人员认为保留一些父母的表观遗传标记可能会增加后代存活的机会。这个新知识的一个影响就是,未来的父母需要考虑他们的行为(饮食,吸烟,喝酒等)对未来出生孩子可能产生的影响。
基因是决定一个人患癌症风险的一个主要因素。具有某些基因突变的人一生中患癌症的风险相对比较高,这些突变通常会遗传给后代。据认知,大约百分之五到百分之十的癌症是有遗传突变引起的。9既然我们知道表观遗传变化也可以遗传,那么在未来的癌症风险研究中,这些类型的变化应该被考虑在内。
癌症中DNA甲基化的变化
癌细胞通常有一个与正常细胞不同的表观基因组或表观遗传学的特征。DNA甲基化程度低于正常水平的DNA被称为低甲基化。甲基化程度高的DNA被称为高甲基化。癌细胞的表观遗传特征是在基因组中甲基化程度的降低(大面积DNA低甲基化)。10 甲基化的降低会影响大量基因的活性。因为甲基化与基因活性的降低有关, 所以甲基化的整体效果是增加基因的活性。如果参与细胞生长的基因甲基化减少,活性将会增加,从而导致细胞分裂,就可能导致癌症的发生。
虽然癌细胞的DNA最常见的是低甲基化,但也有可能是相反的。11 癌细胞的DNA甲基化通常局限于非常特殊的区域(“热点”)。受癌症影响的部位各有不同的类型。DNA甲基化增加的影响与低甲基化相反。高甲基化基因往往表现出活性下降。肿瘤细胞中的DNA甲基化经常出现在肿瘤抑制基因中,这些基因的功能是修复DNA并且控制细胞分裂。当肿瘤抑制基因因为甲基化增加而沉默时,其活性的降低会导致癌症的发生。
这些微小的变化如何影响癌症的生长? 具有高甲基化肿瘤抑制基因的细胞可能比正常甲基化肿瘤抑制基因的细胞生长更快。然而,这并不能解释为什么在某些癌细胞中发现了特定序列的高甲基化,而另一些却没有。这背后确切的原因和机制尚不清楚,但这可能与细胞群体如何随时间进化有关。很可能是不同细胞中的许多基因都发生了甲基化。这些变化中一部分会给这些细胞带来优势,这些细胞能够更快地繁殖,并且占据整个种群。12 一个很好的例子是BRCA1,一个与乳腺癌有关的基因。它在乳腺和卵巢肿瘤中经常被发现过度甲基化,但在其他类型的肿瘤却没有发现甲基化。13
癌症中的组蛋白修饰
组蛋白表观遗传修饰的改变在癌症中起到了重要的作用。如前所诉,这些蛋白质的修饰改变了组蛋白和DNA之间的相互作用。这改变了DNA组蛋白复合物(核小体)的形状,并改变了其他蛋白质与DNA相互作用的方式。
癌细胞的表观基因组通常用组蛋白乙酰化标记物的丢失而作记号,这是由于组蛋白去乙酰化的增加。组蛋白脱乙酰化的过程是由组蛋白脱乙酰基酶或HDACs来催化。正如所料,在不同类型的癌细胞中发现HDACs活性的增加,使HDACs成为表观遗传癌症治疗的重要靶点。14
组蛋白甲基化也被发现会影响癌细胞。组蛋白甲基转移酶(HMTs)是将甲基加到组蛋白的酶,而组蛋白脱甲基酶(HDMs)具有相反的功能。在癌细胞中HMT可能会发生改变,使它们将甲基组的位置放错,这通常会导致抑癌基因沉默。同样地,HDMs的活性也会受到影响。如上所诉,组蛋白甲基化非常复杂。甲基化对基因活性的影响取决于受影响的特定氨基酸。因此,组蛋白上的甲基标记根据其对基因活性的影响氛围激活或者抑制。另一个复杂的问题是,一些HDM已经被发现能够去除激活和抑制标记。这对开发针对HDMs的表观遗传疗法的人提出了一个挑战--必须充分了解它们的功能,才能知道药物将如何影响癌细胞。
肿瘤转移的表观遗传学
癌症的转移是指原发性肿瘤扩散到身体的远处。这种转移是一个多步骤的过程:细胞要与原先肿瘤分开,通过血管到达新的部位,最终在另一个部位建立肿瘤。蛋白质已经被确定可以阻止癌症的扩散。这些转移抑制因子,可以抑制转移过程中的任何步骤。转移癌细胞已被证明在表观遗传学上沉默转移抑制因子,通常是通过这些基因的甲基化。15
癌细胞转移的原因还不完全清楚。比较转移细胞和原发性肿瘤细胞的DNA序列并不能总是能够识别出DNA序列的变化,从而解释细胞之间的差别。2017年,研究人员在至少一个实验模型中发现了转移中的表观遗传学基础。这项研究考查了来自病患的胰腺癌细胞,发现转移细胞的表观基因发生了显著的变化,特别是表观遗传学的变化,影响了参与细胞迁移的基因。16
表观遗传学与癌症预防
与癌症相关的表突变可以预防的因素是环境暴露和我们的行为。消除或减少与致癌化学物质的接触,比如烟草制品中发现的化学物质,可能会减少表突变和相关癌症。其他化学物质和药物也被发现回应起表突变,特别是酒精,它会导致DNA甲基化和组蛋白修饰。17 饮食中富含十字花科蔬菜,如西兰花,卷心菜,甘蓝菜,都可以降低患前列腺癌的风险。18几种植物化学物质在这其中起作用,包括莱菔硫烷和二吲哚甲烷(DIM)。当研究人员将前列腺癌细胞暴露在DIM中时,HDAC活性降低,p21肿瘤抑制蛋白的数量增加。
表观遗传学与癌症检测
目前检测癌症的方法使用成像技术(X射线,PET扫描,超声波等)或直接从可疑区域取样来检测肿瘤生长。这些方法在检测癌症方面都能取到很大的作用;然而其中很多方法依赖于对已经存在的异常生长的检测。表观遗传学检测方法正被设计成超越当前检测的灵敏度和特异性。
甲基化特异性 PCR (MSP) 是一种表观遗传学检测方法,用于识别DNA甲基化异常,所以应该有可能识别出癌症特有的模式。这种分析依赖于亚硫酸氢盐测试,一种可以区分已甲基化的胞嘧啶和未发生改变的胞嘧啶的技术。MSP是有用的,因为它的敏感性--它可以检测出成千上万个细胞中的单个肿瘤细胞。19 另一个好处就是,与活检不同,MSP是非侵入性的。MSP可以检测血浆,粪便,痰和尿液样本中的癌症DNA甲基化模式。
液体活检是一种依靠血液样本分析的癌症检查方法。液体活检依赖于血液中肿瘤细胞和/或肿瘤DNA的存在。这些可以用来分析DNA突变和表观遗传的变化。20 液体活检被用于快速增长的疾病,包括癌症。使用液体或组织检查来检测癌症中的表观遗传变化有一些缺点。最重要的是我们对与癌症相关的表突变的知识有限。由于不同类型的癌症与不同的表观突变相关,所以很难设计一种检测癌症的通用检测方法。但由于每一个已经确认的变化都必须在许多患者身上得到证实,这个过程就被延缓了。
表观遗传学相关蛋白作为癌症的生物标记物
生物标志物是一种不易直接测量的疾病指标。生物标志物的例子包括使用血胆固醇水平和血压作为心脏病的指标。目前对表观遗传癌症检测方法的研究大多依赖于识别和检测特定的表观遗传变化。理想情况下,最好能找到一组在大多数或所有类型癌症中都存在的变化(或蛋白质)。2017年的一项研究已经确定了一种可以作为这种标记物的蛋白质。UHRF1是一种由UHRF1基因编制的蛋白质。它与特定的DNA、序列结合,在哪里它招募DNMT1,一种DNA甲基转移酶。它还可以协调不同的表观遗传酶,如DNMTs和HDAC是,从而维持正常的DNA甲基化模式和组蛋白修饰模式。21UHR1在多种癌症中表现出高度的活性,使其成为一种潜在的通用癌症生物标志物。22 UHRF1被鉴定为一种促进癌症发展的蛋白质。UHRF1水平的增加通过破坏DNMTs的稳定性导致DNA的整体低甲基化。在不同类型的癌症中,UHRF1活性的增加与肿瘤的生长,转移,和对放疗的抵抗有关。包括肺癌,肝癌,乳腺癌,胰腺癌,结直肠癌,前列腺癌和肾癌。太多的UHRF1会导致较低的生存率,更高的抗药性,以及更高的复发率。在肿瘤发生的早期就可以检测到异常的UHRF1的活性,是肿瘤早期诊断的一个好选择。靶向UHRF1也可以提高放疗的有效性。
针对表观遗传变化的癌症治疗
目前大多数癌症治疗,包括放疗和化疗,都是细胞毒性治疗。他们的目标是杀死癌细胞。然而,这些治疗方法的有效性有限,因为他们经常损坏或者会杀死正常的细胞,而且癌细胞中经常会产生抗药性。由于这些局限性,其他类型的治疗正在研究中。包括免疫疗法和表观遗传治疗。
表观遗传治疗或者“epi药物”是指一种可以逆转癌细胞异常表观遗传活动的药物。epi药物的主要好处是它们没有毒性,它们的目标就是“重新编制”癌细胞。23 与基因突变不同的是,表突变是可逆的,这为逆转癌细胞的表突变提供了可能。表观遗传治疗的靶点是参与表观遗传修饰的酶,包括HDACs,DNMTs和HDMs。尽管任何类型的表观遗传酶都是表观遗传治疗潜在的靶点,但研究几种在那些不太复杂而且研究得很好的酶上面。目前正在研究中的两种主要epi药物是HDAC抑制剂和DNMT抑制剂。
组蛋白去乙酰化酶(HDAC)抑制剂
组蛋白乙酰化是酶向组蛋白中添加乙酰基的过程。结果常常是基因活性的增加。相反的过程,组蛋白去乙酰化,则会导致基因沉默。在许多类型的癌细胞中,组蛋白去乙酰基酶的活性很高。这种增加与肿瘤抑制基本和DNA修复基因的沉默有关。HDAC抑制剂的目的是降低HDAC的活性,间接提高肿瘤抑制因子的活性。
目前正在研究许多HDAC抑制剂,其中一些已被批准用于临床,包括伏立诺斯塔(Zolinza®),罗米地平(Istodax®),贝利诺斯特(Beleodaq)以及帕诺比诺司他(Farydak®)。24 这些epi药物也有一些局限性和缺点。其中之一就是这些药物只对治疗血液(血液)癌有效,包括淋巴瘤,白血病和骨髓瘤。另一个缺点是这些药物有副作用,包括疲劳和腹泻。它们对骨髓也有毒性,并会减少血细胞的数量。副作用其中的原因是缺乏特性。针对所有类型的HDAC(或其他酶)而引起副作用。HDAC抑制剂是专门针对研究的很好的HDAC而设计的,可以有更好的效果减少副作用。
DNA甲基转移酶(DNMT)抑制剂
如上所诉,DNA甲基化是一种称为DNA甲基转移酶的酶在DNA的碱基上添加甲基的过程。因为这些变化会影响基因的活性,所以DNA甲基化在癌症的发展中骑着重要的作用。肿瘤抑制基因通常会通过高度的甲基化以及沉默。因此DNMT抑制剂已被研究作为有可能的epi药物。25 理论上,DNMT抑制剂可以逆转基因沉默,恢复正常的抑癌功能。
两种DNMT抑制剂已被证明是有效的抗癌药物,目前被批准用于治疗急性髓系白血病和骨髓增长综合症:氮胞苷(Vidaza®)和地西塔(Dacogen®)。但这些药物进入细胞时,它们与DNA相互作用,抑制任何产生的DNMT。结合的DNMT不能使DNA的其他区域进一步甲基化,最终被破坏。尽管这两种免疫药物在临床试验上都有广泛的应用,但他们都有一些缺点。一个缺点是它们的化学不稳定性;它们很快会被分解,通常不到 一个小时,药物就会变成没有活性的化合物。另一个缺点是药物的毒性。它们会导致DNA损伤和免疫功能下降。
一种新的实验性DNMT抑制剂西布林已被证明是有效的。虽然它还没有达到临床试验阶段,但他是有希望的,因为它可以改变氮胞苷(Vidaza®)和地西塔(Dacogen®)的缺点。斑马碱可以抑制DNMTs并逆转癌细胞的基因沉默。他的作用和氮胞苷与地西塔类似,是通过与DNA的相互作用。26 当DNMTs遇到粘在其上的斑马碱的DNA时,会形成一种非常稳定的复合物,“捕获”DNMT“并阻止甲基化。与两种经批准的DNMT抑制剂相比,西布林具有一些优点。它在体内更稳定,而且毒性更小。
联合表观遗传药物治疗
癌症等复杂疾病往往难以治疗。癌症也经常对任何个体治疗产生耐药性。联合治疗已成为一种更有效地治疗癌症的方法。联合治疗是指同时使用多种治疗。对于癌症患者来说,这通常涉及手术,表观遗传药物,放疗,化疗和靶向治疗的一些组合。
表观遗传药物和放疗/化疗的结合似乎是有用的,因为每种疗法都可以弥补另一种疗法的缺点。放疗和化疗可以有效地杀死癌细胞并减缓生长;然而,这些治疗方法本身往往是不够的。如果没有杀死所有的癌细胞,那么复发的可能性是非常大的。包括表观遗传药物在内的联合治疗可能会增加化疗和放疗的敏感性。 27
联合表观遗传学治疗也是一种可能。这包括使用两种或两种以上不同类型的表观遗传药物,包括HDAC抑制剂和DNMT抑制剂。联合表观遗传学治疗的一个显著好处是它允许使用较低剂量的药物。这可以减少副作用,提高疗效。对急性髓系白血病细胞的研究表明,联合治疗中运用的昔布他滨治疗可导致癌细胞死亡增加。28 与单独使用HDAC抑制剂相比,联合治疗使用HDAC和DNMT抑制剂可以导致组蛋白乙酰化增加。这种增加可以解释为DNA甲基化和组蛋白修饰中间提供“通讯”。组蛋白修饰增强了DNA甲基化的作用。让DNA上甲基结合的蛋白质有能力吸收其他酶,包括HDACs,进一步降低基因活性。29
局限性和未来方向
目前的表观遗传学治疗是有希望的;然而,要使其更有效,必须克服几个障碍。虽然表观遗传药物已被证明对血液系统肿瘤有效,但其主要局限性是对实体瘤无效。这在很大程度上是因为肿瘤的环境中氧含量很低(低氧=缺氧)。30 肿瘤中由于存在的血管而发生缺氧,血管不能为快速生长的癌细胞提供足够的氧气。这种情况会改变肿瘤细胞的行为。缺氧会导致细胞产生一组不同的酶和蛋白质,这些酶和蛋白质控制着基因的活性。此外,生活在缺氧条件下的肿瘤细胞具有异常的表观遗传特征,其特征是活性组蛋白标记减少,抑制组蛋白标记增加,组蛋白乙酰化和DNA甲基化减少。这一点很重要,因为目前的HDAC和DNMT抑制剂似乎对缺氧条件下生长的肿瘤细胞无效。为了更好地了解缺氧条件下癌细胞的表观遗传模式,需要进行研究。这些信息将有助于我们开发针对实体瘤的表观遗传药物。
联合治疗也可以改进。这种治疗方法依赖于不同类型的癌症疗法的协同。然而,这还需要进行广泛的研究,以发现不同药物的最佳组合,适当的剂量和时机。开发新的表观遗传药物对改善癌症治疗也至关重要。其中一个主要目标是涉及更具特异性的药物。正在进行的研究包括进一步探索与癌症相关的表观遗传生物标志物,并致力于了解癌细胞耐药的表观遗传机制。研究癌症表观遗传学的一个主要原因是为开发安全,有效的治疗方法以及帮助预防和检测癌症提供信息。
- 1 Chen, Q. W., Zhu, X. Y., Li, Y. Y., & Meng, Z. Q. (2014). Epigenetic regulation and cancer. Oncology Reports, 31, 523-532. [PUBMED]
- 2ab Kouzarides, T. (2007). Chromatin Modifications and Their Function. Cell, 128, 693-705. [PUBMED]
- 3 Annunziato, A. (2008) DNA Packaging: Nucleosomes and Chromatin. Nature Education 1(1):26. [View]
- 4 Bannister, A. J., & Kouzarides T. (2011). Regulation of chromatin by histone modifications. Cell, 21(3), 381-395. [PUBMED]
- 5 Powledge, T. M. (2014, August 12). How much of human DNA is doing something? Retrieved July 26, 2017 [View]
- 6 Joehanes, R., et al. (2016). Epigenetic Signatures of Cigarette Smoking. Circulation: Cardiovascular Genetics, 9(5), 436-447. [PUBMED]
- 7 Dias, B. G., & Ressler, K. J. (2013). Parental olfactory experience influences behavior and neural structure in subsequent generations. Nature Neuroscience, 17, 89-96. [PUBMED]
- 8ab Heard, E., & Martienssen, R. A. (2014). Transgenerational Epigenetic Inheritance: myths and mechanisms. Cell, 157(1), 95-109. [PUBMED]
- 9 Genetic Testing for Hereditary Cancer Syndromes. (2013, April 11). Retrieved July 14, 2017. [View]
- 10 Sharma, S., Kelly, T. K., Jones, & P. A. (2010). Epigenetics in cancer. Carcinogenesis, 31(1), 27-36. [PUBMED]
- 11 De Bustros, A., Nelkin, B. B., Silverman, A., Ehrlich, G., Poiesz, B., & Baylin, S. B. (1988). The short arm of chromosome 11 is a "hot spot" for hypermethylation in human neoplasia. Proc Natl Acad Sci USA, 85(15), 5693-5697. [PUBMED]
- 12 Esteller, M. (2002). CpG island hypermethylation and tumor suppressor genes: a booming present, a brighter future. Oncogene, 21(35), 5427-5440. [PUBMED]
- 13 Esteller, M., et al. (2000). Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J Natl Cancer Inst, 92(7), 564-569. [PUBMED]
- 14 Halkidou,K., et al. (2004). Upregulation and nuclear recruitment of HDAC1 in hormone refractory prostate cancer. Prostate, 59, 177–189. [PUBMED]
- 15 Li, Q. & Chen, H. (2011). Epigenetic modifications of metastasis suppressor genes in colon cancer metastasis. Epigenetics, 6(7), 849-852. [PUBMED]
- 16 McDonald, O. G. et al. (2017). Epigenetic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nature Genetics, 49, 367-376. [PUBMED]
- 17 Mahnke, A. H., Miranda, R. C., & Homanics, G. E. (2017). Epigenetic mediators and consequences of excessive alcohol consumption. Alcohol, 60, 1-6. [PUBMED]
- 18 Beaver, L. M., et al. (2012). 3,3′-Diindolylmethane, but not indole-3-carbinol, inhibits histone deacetylase activity in prostate cancer cells. Toxicology and Applied Pharmacology, 263(3), 345-351. [PUBMED]
- 19 Zhu, J., & Yao, X. (2008). Use of DNA methylation for cancer detection: Promises and challenges. The International Journal of Biochemistry and Cell Biology, 41, 147-154. [PUBMED]
- 20 Alix-Panabières, C. & Pantel, K. (2013). Circulating Tumor Cells: Liquid Biopsy of Cancer. Clinical Chemistry, 59(1), 110-118. [PUBMED]
- 21 Unoki, M., Brunet, J., & Mousli, M. (2009). Drug discovery targeting epigenetic codes: the great potential of UHRF1, which links DNA methylation and histone modifications, as a drug target in cancers and toxoplasmosis. Biochem Pharmacol, 78, 1279-1288. [PUBMED]
- 22 Ashraf, W. et al. (2017). The epigenetic integrator UHRF1: on the road to become a universal biomarker for cancer. Oncotarget. [PUBMED]
- 23 Ronnekleiv-Kelly, S. M., Sharma, A., & Ahuja, N. (2017) Epigenetic therapy and chemosensitization in solid malignancy. Cancer Treatment Reviews, 55, 200-208. [PUBMED]
- 24 Ceccacci, E., & Minucci, S. (2016). Inhibition of histone deacetylases in cancer therapy: lessons from leukaemia. British Journal of Cancer, 114(6), 605-611. [PUBMED]
- 25 Gnyszka, A., Jastrzebski, Z., & Flis, S. (2013). DNA Methyltransferase Inhibitors and Their Emerging Role in Epigenetic Therapy of Cancer. Anticancer Research, 33(8), 2989-2996. [PUBMED]
- 26 Champion, C. et al. (2010). Mechanistic Insights on the Inhibition of C5 DNA Methyltransferases by Zebularine. PLoS One, 5(8), e12388. [PUBMED]
- 27 Pajonk, F., Vlashi, E., & McBride, W. H. (2010). Radiation Resistance of Cancer Stem Cells: The 4 R’s of Radiobiology Revisited. Stem Cells, 28(4), 639–648. [PUBMED]
- 28 Young, C. S., Clarke, K. M., Kettyle, L. M., Thompson, A., & Mills, K. I. (2017). Decitabine-Vorinostat combination treatment in acute myeloid leukemia activates pathways with potential for novel triple therapy. Oncotarget. [PUBMED]
- 29 Jones, P. L., et al. (1998). Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nature Genetics, 19, 187-191. [PUBMED]
- 30 Ramachandran, S., Ient, J., Göttgens, E., Krieg, A. J., & Hammond, E. M. (2015) Epigenetic Therapy for Solid Tumors: Highlighting the Impact of Tumor Hypoxia. Genes, 6, 935-956. [PUBMED]